| |
|
Ahn,S., J.A. Anderson, M.E.
Sorrells, and S. Tanksley. 1993. Homoeologous relationships
of rice, wheat, and maize chromosomes. Mol. Gen. Genet.
241: 483-490.
Ahn, S. and S. Tanksley. 1993.
Comparative linkage maps of the rice and maize genomes. Proc.
Natl. Acad. Sci. (USA) 90:7980-7984.
Altschul, S.F., W. Gish, W.
Miller, E.W. Myers, and D.J. Lipman. 1990. Basic local alignment
search tool. J. Mol. Biol 215:403-410.
Barry, G.F. 2001. The use of
the Monsanto draft rice genome sequence in research. Plant
Physiology 125:1164-1165.
Bastide, M.d.l., D. Johnson,
V. Balija, and W.R.McCombie. 2001. Strategies and techniques
for finishing genomic sequence. In Rice Genetics IV
(eds. G.S. Khush D.S. Brar, and B. Hardy). The International
Rice Research Institute Press, Manila.
Birney, E. and R. Durbin. 2000.
Using GeneWise in the Drosophila annotation experiment. Genome
Research 10: 547-548.
Cai, H.W. and H. Morishima.
2000. Genomic regions affecting seed shattering and seed dormancy
in rice. Theoretical and Applied Genetics 100:
840-846.
Chen, M., G. Presting, W. Barbazuk,
JLGoicoechea, B. Blackmon, G. Fang, H. Kim, D. Frisch, Y.
Yu, S. Higingbottom, K. Phimphilai, S. Phimphilai, S. Thurmond,
B. Gaudette, P. Li, J. Liu, J. Hatfield, D. Main, S. Sun,
K. Farrar, C. Henderson, L. Barnett, R. Costa, B. Williams,
S. Walser, M. Atkins, C. Hall, I. Bancroft, J. Salse, F. Regad,
T. Mohapatra, N. Singh, A. Tyagi, C. Soderlund, R. Dean, and
R. Wing. 2002. An Integrated Physical and Genetic Map of the
Rice Genome. Plant Cell 14: 537-545.
Cheng, Z., C. Buell, R. Wing,
M. Gu, and J. Jiang. 2001a. Toward a Cytological Characterization
of the Rice Genome. Genome Research 11: 2133-2141.
Cheng, Z., G. Presting, C.R.
Buell, and R.A. Wing. 2001b. High resolution pachytene chromosome
mapping of bacterial artificial chromosomes anchroed by genetic
markers reveals the centromere location and the distribution
of genetic recombination along chromosome 10 of rice. Genetics
157: 1749-1757.
Clamp, M., D. Andrews, D. Barker,
P. Bevan, G. Cameron, Y. Chen, L. Clark, T. Cox, J. Cuff,
V. Curwen, T. Down, R. Durbin, E. Eyras, J. Gilbert, M. Hammond,
T. Hubbard, A. Kasprzyk, D. Keefe, H. Lehvaslaiho, V. Iyer,
C. Melsopp, E. Mongin, R. Pettett, S. Potter, A. Rust, E.
Schmidt, S. Searle, G. Slater, J. Smith, W. Spooner, A. Stabenau,
J. Stalker, E. Stupka, A. Ureta-Vidal, I. Vastrik, and E.
Birney. 2003. Ensembl 2002: accommodating comparative genomics.
Nucleic Acids Research 31: 38-42.
Coe, E., K. Cone, M. McMullen,
S. Chen, G. Davis, J. Gardiner, E. Liscum, M. Polacco, A.
Paterson, H. Sanchez-Villeda, C. Soderlund, and R.A. Wing.
2002. Access to the maize genome: an integrated physical and
genetic map. Plant Physiology 128: 9-12.
Cone, K., M. McMullen, I.V.
Bi, G. Davis, Y. Yim, J. Gardiner, M. Polacco, H. Sanchez-Villeda,
Z. Fang, S. Schroeder, S. Havermann, J. Bowers, A. Paterson,
C. Soderlund, F. Engler, R.A. Wing, and E. Coe. 2002. Genetic,
Physical, and Informatics Resources for Maize. On the Road
to an Integrated Map. Plant Physiololgy 130:
1598-1605.
Davenport, R.J. 2001. Syngenta
Finishes. Consortium Goes On 291: 807.
Devos, K.M., J.K.M. Brown,
and J.L. Bennetzen. 2002. Genome size reduction through illegitimate
recombination counteracts genome expansion in Arabidopsis.
Genome Research 12: 1075-1079.
Ding, Y., M.D. Johnson, W.Q.
Chen, D. Wong, Y.J. Chen, S.C. Benson, J.Y. Lam, Y.M. Kim,
and H. Shizuya. 2001. Five-color-based high-information-content
fingerprinting of bacterial artificial chromosome clones using
type IIS restriction endonucleases. Genomics 74:
142-154.
Dowell, R.D., R.M. Jokerst,
A. Day, S.R. Eddy, and L. Stein. 2001. The Distributed Annotation
System. BMC Bioinformatics 2: 7.
Ewing, B. and P. Green. 1998.
Base-calling of automated sequencer traces using phred. II.
Error probabilities. Genome Res. 8: 186-194.
Ewing, B., L. Hillier, M.
Wendl, and P. Green. 1998. Base-Calling of Automated Sequencer
Traces Using Phred. I. Accuracy Assessment. Genome
Res. 8: 175-185.
Feng, Q., Y. Zhang, P. Hao,
S. Wang, G. Fu, Y. Huang, Y. Li, J. Zhu, Y. Liu, X. Hu, P.
Jia, Q. Zhao, K. Ying, S. Yu, Y. Tang, Q. Weng, L. Zhang,
Y. Lu, J. Mu, L.S. Zhang, Z. Yu, D. Fan, X. Liu, T. Lu, C.
Li, Y. Wu, T. Sun, H. Lei, T. Li, H. Hu, J. Guan, M. Wu, R.
Zhang, B. Zhou, Z. Chen, L. Chen, Z. Jin, R. Wang, H. Yin,
Z. Cai, S. Ren, G. Lv, W. Gu, G. Zhu, Y. Tu, J. Jia, J. Chen,
H. Kang, X. Chen, C. Shao, Y. Sun, Q. Hu, X. Zhang, W. Zhang,
L. Wang, C. Ding, H. Sheng, J. Gu, S. Chen, L. Ni, F. Zhu,
W. Chen, L. Lan, Y. Lai, Z. Cheng, M. Gu, J. Jiang, J. Li,
G. Hong, Y. Xue, and B. Han. 2002. Sequence and analysis of
rice chromosome 4. Nature 420: 316-320.
Flicek, P., E. Keibler, P.
Hu, I. Korf, and M.R. Brent. 2003. Leveraging the mouse genome
for gene prediction in human: from whole- genome shotgun reads
to a global synteny map. Genome Res 13: 46-54.
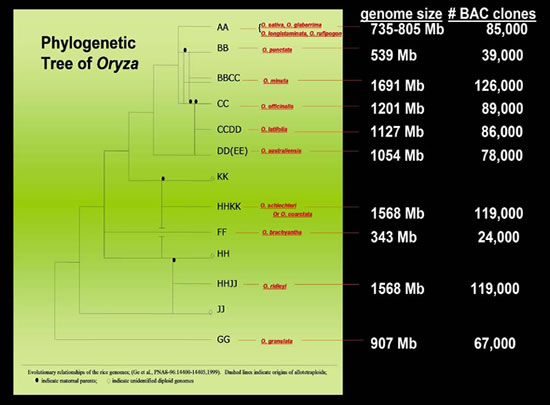
Ge, S., T. Sang, B.-R. Lu,
and D.-Y. Hong. 1999. Phylogeny of rice genomes with emphasis
on origins of allotetraploid species. Proc Natl Acad Sci
U S A 96: 14400-14405.
Goff, S.A., D. Ricke, T.H.
Lan, G. Presting, R. Wang, M. Dunn, J. Glazebrook, A. Sessions,
P. Oeller, H. Varma, D. Hadley, D. Hutchison, C. Martin, F.
Katagiri, B.M. Lange, T. Moughamer, Y. Xia, P. Budworth, J.
Zhong, T. Miguel, U. Paszkowski, S. Zhang, M. Colbert, W.L.
Sun, L. Chen, B. Cooper, S. Park, T.C. Wood, L. Mao, P. Quail,
R. Wing, R. Dean, Y. Yu, A. Zharkikh, R. Shen, S. Sahasrabudhe,
A. Thomas, R. Cannings, A. Gutin, D. Pruss, J. Reid, S. Tavtigian,
J. Mitchell, G. Eldredge, T. Scholl, R.M. Miller, S. Bhatnagar,
N. Adey, T. Rubano, N. Tusneem, R. Robinson, J. Feldhaus,
T. Macalma, A. Oliphant, and S. Briggs. 2002. A draft sequence
of the rice genome (Oryza sativa L. ssp. japonica). Science
296: 92-100.
Green, P. 1999. swat/cross_match/phrap
package. P. Green.
Hass, B., J.W. Lilly, J.C.
Pires, R. Porter, P.L. Philips, and S.A. Jackson. 2003. Comparative
genetics at the gene and chromosome levels between rice (Oryza
sativa) and wild rice (Zizania palustris). Theoretical
and Applied Genetics (in press).
Hulbert, S.H., T.E. Richter,
J.B. Axtell, and J.L. Bennetzen. 1990. Genetic mapping and
characterization of sorghum and related crops by means of
maize DNA probes. Proc. Natl. Acad. Sci. (USA) 87:4251-4255.
IRGSP. 2002. Completion of
the rice genome sequence announced.
Jackson, S.A., M.L. Wang,
H.M. Goodman, and J. Jiang. 1998. Application of fiber-FISH
in physical mapping of Arabidopsis thaliana. Genome
41: 566-572.
Jaiswal, P., D. Ware, J. Ni,
K. Chang, W. Zhao, S.Schmidt, X. Pan, K. Clark, L. Teytelman,
S. Cartinhour, and L. Stein. 2002. Gramene: development and
integration of trait and gene ontologies for rice. Comparative
and Functional Genomics 3: 2.
Jamison, D.C., J.W. Thomas,
and E.D. Green. 2000. ComboScreen facilitates the multiplex
hybridization-based screening of high-density clone arrays.
Bioinformatics 16: 678-684.
Korf, I., P. Flicek, D. Duan,
and M.R. Brent. 2001. Integrating genomic homology into gene
structure prediction. Bioinformatics 17: S140-148.
Kurata, N., G. Moore, Y. Nagamura,
T. Foote, M. Yano, Y. Minobe, and M.D. Gale. 1994. Conservation
of Genome Structure between Rice and Wheat. Bio/Technology
12: 276-278.
Mao, L., T.C. Wood, Y. Yu,
M.H. Budiman, S.S. Woo, M. Sasinowski, S.A. Goff, R.A. Dean,
and R.A. Wing. 2000. Rice Transposable Elements: A Survey
of 73,000 Sequence-Tagged-Connectors (STCs). Genome Research
10: 982-990.
Marra, M., T. Kkucaba, N.
Dietrich, E. Green, B. Brownstein, R. Wilson, K. McDonald,
L. Hillier, J. McPherson, and R. Waterston. 1997. High throughput
fingerprint analysis of large-insert clones. Genome Research
7: 1072-1084.
McCouch, S.R., L. Teytelma,
Y. Xu, K.B. Lobos, K. Clare, M. Walton, B. Fu, R. Magnirang,
Z. Li, Y. Xing, Q. Zhang, I. Kono, M. Yano, R. Fjellstrom,
G. DeClerck, D. Schneider, S. Cartinhour, D. Ware, and L.
Stein. 2002. Development of 2,243 new SSR markers for rice
(Oryza sativa L.). DNA Research (in press).
Moore, G., K.M. Devos, Z.
Wang, and M.D. Gale. 1995. Grasses, line up and form circle.
Curr.Biol. 5: 737-739.
Mulder, N., R. Apweiler, T.
Attwood, A. Bairoch, D. Barrell, A. Bateman, D. Binns, M.
Biswas, P. Bradley, P. Bork, P. Bucher, R. Copley, E. Courcelle,
U. Das, R. Durbin, L. Falquet, W. Fleischmann, S. Griffiths-Jones,
D. Haft, N. Harte, N. Hulo, D. Kahn, A. Kanapin, M. Krestyaninova,
R. Lopez, I. Letunic, D. Lonsdale, V. Silventoinen, S. Orchard,
M. Pagni, D. Peyruc, C. Ponting, J. Selengut, F. Servant,
C. Sigrist, R. Vaughan, and E. Zdobnov. 2003. The InterPro
Database, 2003 brings increased coverage and new features.
Nucleic Acids Research 31: 315-318.
Mullikin, J. and Z. Ning.
2003. The phusion assembler. Genome Research 13:
81-90.
O'Brien, S.J., J. Wienberg,
and L.A. Lyons. 1997. Comparative genomics: lessons from cats.
Trends Genet 13: 393-399.
Rivas, E. and S.R. Eddy. 2001.
Noncoding RNA gene detection using comparative sequence analysis.
BMC Bioinformatics 2: 8.
Rozen, S. and H. Skaletsky.
2000. Primer3 on the WWW for general users and for biologist
programmers. Methods Mol Biol. 132: 365-386.
Salamov, A. and V. Solovyev.
2000. Ab initio gene finding in Drosophila genomic DNA. Genome
Research 10: 516-522.
SanMiguel, P., B.S. Gaut,
A. Tikhonov, Y. Nakajima, and J.L. Bennetzen. 1998. The paleontology
of intergene retrotransposons of maize. Nat Genet 20:
43-45.
SanMiguel, P.J., W. Ramakrishna,
J.L. Bennetzen, C. Busso, and J. Dubcovsky. 2002. Transposable
elements, genes and recombination in a 215-kb contig from
wheat chromosome 5Am. Functional and Integrative Genomics
2: 70-80.
Sasaki, T. and B. Burr. 2000.
International Rice Genome Sequencing Project: the effort to
completely sequence the rice genome. Curr Opin Plant Biol
3: 138-141.
Sasaki, T., T. Matsumoto,
K. Yamamoto, K. Sakata, T. Baba, Y. Katayose, J. Wu, Y. Niimura,
Z. Cheng, Y. Nagamura, B.A. Antonio, H. Kanamori, S. Hosokawa,
M. Masukawa, K. Arikawa, Y. Chiden, M. Hayashi, M. Okamoto,
T. Ando, H. Aoki, K. Arita, M. Hamada, C. Harada, S. Hijishita,
M. Honda, Y. Ichikawa, A. Idonuma, M. Iijima, M. Ikeda, M.
Ikeno, S. Ito, T. Ito, Y. Ito, A. Iwabuchi, K. Kamiya, W.
Karasawa, S. Katagiri, A. Kikuta, N. Kobayashi, I. Kono, K.
Machita, T. Maehara, H. Mizuno, T. Mizubayashi, Y. Mukai,
H. Nagasaki, M. Nakashima, Y. Nakama, Y. Nakamichi, M. Nakamura,
N. Namiki, M. Negishi, I. Ohta, N. Ono, S. Saji, K. Sakai,
M. Shibata, T. Shimokawa, A. Shomura, J. Song, Y. Takazaki,
K. Terasawa, K. Tsuji, K. Waki, H. Yamagata, H. Yamane, S.
Yoshiki, R. Yoshihara, K. Yukawa, H. Zhong, H. Iwama, T. Endo,
H. Ito, J.H. Hahn, H.I. Kim, M.Y. Eun, M. Yano, J. Jiang,
and T. Gojobori. 2002. The genome sequence and structure of
rice chromosome 1. Nature 420: 312-316.
Shantz. 1954. The place of
grasslands in the earth's cover of vegetation. Ecology
35: 143-145.
Shishido, R., Y. Sano, and
K. Fukui. 2001. Ribosomal DNAs: an exception to the conservation
of gene order in rice genomes. Mol. Gen. Genet. 263:
586-591.
Soderlund, C., F. Engler,
J. Hatfield, S. Blundy, M. Chen, Y. Yu, and R.A. Wing. 2002.
Mapping Sequence to FPC Rice Map. In Computational Biology
and Genome Informatics. (eds. C. Wu P. Wang, and J. Wang),
pp. In press. World Scientific Publishing.
Soderlund, C., I. Longden,
and R. Mott. 1997. FPC: A system for building contigs from
restriction fingerprinted clones. CABIOS 13:
523-535.
Thomas, J.W., A.B. Prasad,
T.J. Summers, S.Q. Lee-Lin, V.V. Maduro, J.R. Idol, J.F. Ryan,
P.J. Thomas, J.C. McDowell, and E.D. Green. 2002. Parallel
construction of orthologous sequence-ready clone contig maps
in multiple species. Genome Res 12: 1277-1285.
Tikhonov, A.P., P.J. SanMiguel,
Y. Nakajima, N.M. Gorenstein, J.L. Bennetzen, and Z. Avramova.
1999. Colinearity and its exceptions in orthologous adh regions
of maize and sorghum. Proc Natl Acad Sci U S A 96:
7409-7414.
Tomkins, J.P., G. Davis, D.
Main, Y. Yim, N. Duru, T. Musket, J.L. Goicoechea, D.A. Frisch,
E.H. Coe, Jr., and R.A. Wing. 2002. Construction and Characterization
of a Deep-Coverage Bacterial Artificial Chromosome Library
for Maize. Crop Sci 42: 928-933.
Uozu, S., H. Ikehashi, N.
Ohmido, H. Ohtsubo, E. Ohtsubo, and K. Fukui. 1997. Repetitive
sequences: cause for variation in genome size and chromosome
morphology in the genus Oryza. Plant Mol Biol 35:
791-799.
Vaughan, D.A. 1994. The
wild relatives of rice. International Rice Research Institute,Manila.
Ware, D., P. Jaiswal, J. Ni,
X. Pan, K. Chang, K. Clark, L. Teytelman, S. Schmidt, W. Zhao,
S. Cartinhour, S. McCouch, and L. Stein. 2002a. Gramene: a
resource for comparative grass genomics. Nucleic Acids
Res 30: 103-105.
Ware, D.H., P. Jaiswal, J.
Ni, I.V. Yap, X. Pan, K.Y. Clark, L. Teytelman, S.C. Schmidt,
W. Zhao, K. Chang, S. Cartinhour, L.D. Stein, and S.R. McCouch.
2002b. Gramene, a tool for grass genomics. Plant Physiol
130: 1606-1613.
Wheeler, D., DMChurch, S.
Federhen, A. Lash, T. Madden, J. Pontius, G. Schuler, L. Schriml,
E. Sequeira, T. Tatusova, and L. Wagner. 2003. Database resources
of the National Center for Biotechnology. Nucleic Acids
Research 31: 28-33.
Wing, R.A., Y. Yu, G. Presting,
D.A. Frisch, T.C. Wood, S.S. Woo, M.H. Budiman, L. Mao, H.R.
Kim, T. Rambo, E. Fang, B. Blackmon, J.L. Goicoechea, S. Higingbottom,
M. Sasinowski, J.P. Tomkins, R.A. Dean, and C.A. Soderlund.
2001. Sequence-tagged connector/DNA fingerprint framework
for rice genome sequencing. In Rice Genetics IV (eds.
G.S. Khush D.S. Brar, and B. Hardy), pp. 215-225. The International
Rice Research Institute Press, Manila.
Wing, R.A., Y. Yu, T. Rambo,
J. Currie, C. Saski, H.R. Kim, K. Collura, S. Thompson, J.
Simmons, T.J. Yang, G.N. Park, A.J. Patel, S. Thurmond, D.
Henry, R. Oates, M. Palmer, G. Pries, J. Gibson, H. Anderson,
M. Paradkar, L. Crane, J. Dale, M. Carver, T. Wood, D. Frisch,
F. Engler, C. Soderlund, W.R.M.e. al., P. Minx, D. Johnson,
H. Cordum, E. Mardis, R. Wilson, J. Messing, R. Song, G. Fuks,
V. Llaca, S. Kovchak, S. Young, Z. Cheng, J. Jiang, M.A. Johns,
L. Mao, H. Pan, R.A. Dean, J.E. Bowers, A.H. Paterson, Q.
Yuan, S. Ouyang, J. Liu, K.M. Jones, K. Gansberger, K. Moffat,
J. Hill, T. Tsitrin, L. Overton, J. Bera, M. Kim, S. Jin,
L. Tallon, A. Ciecko, G. Pai, S.V. Aken, T. Utterback, S.
Reidmuller, J. Bormann, T. Feldblyum, J. Hsiao, V. Zismann,
S. Blunt, A.d. Vazeilles, T. Shaffer, H. Koo, B. Suh, Q. Yang,
B. Haas, J. Peterson, M. Pertea, N. Volfovsky, J. Wortman,
O. White, S.L. Salzberg, C.M. Fraser, and C.R. Buell. 2003.
Sequence, annotation, and comparative analyses of rice chromosome
10. (Submitted) XX: XX-XX.
Yuan, Q., F. Liang, J. Hsiao,
V. Zismann, M.I. Benito, J. Quackenbush, R. Wing, and R. Buell.
2000. Anchoring of rice BAC clones to the rice genetic map
in silico. Nucleic Acids Res 28: 3636-3641.
Zhao, Q., Y. Zhang, Z. Cheng,
M. Chen, S. Wang, Q. Feng, Y. Huang, Y. Li, Y. Tang, B. Zhou,
Z. Chen, S. Yu, J. Zhu, X. Hu, J. Mu, K. Ying, P. Hao, L.
Zhang, Y. Lu, L.S. Zhang, Y. Liu, Z. Yu, D. Fan, Q. Weng,
L. Chen, T. Lu, X. Liu, P. Jia, T. Sun, Y. Wu, Y. Zhang, Y.
Lu, C. Li, R. Wang, H. Lei, T. Li, H. Hu, M. Wu, R. Zhang,
J. Guan, J. Zhu, G. Fu, M. Gu, G. Hong, Y. Xue, R. Wing, J.
Jiang, and B. Han. 2002. A Fine Physical Map of the Rice Chromosome
4. Genome Research 12: 817-823.
Zhou, Y., W. Li, W. Wu, Q.
Chen, D. Mao, and A.J. Worland. 2001. Genetic dissectionof
heading time and its components in rice. Theoretical and
Applied Genetics 102: 1236-1242.
|